When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how do regulators know it’s safe and effective? The answer lies in something called the 80-125% rule-a quiet but powerful standard that decides whether a generic drug can hit the market. It’s not about how much active ingredient is in the pill. It’s not about price. It’s about what happens inside your body after you take it.
What the 80-125% Rule Actually Means
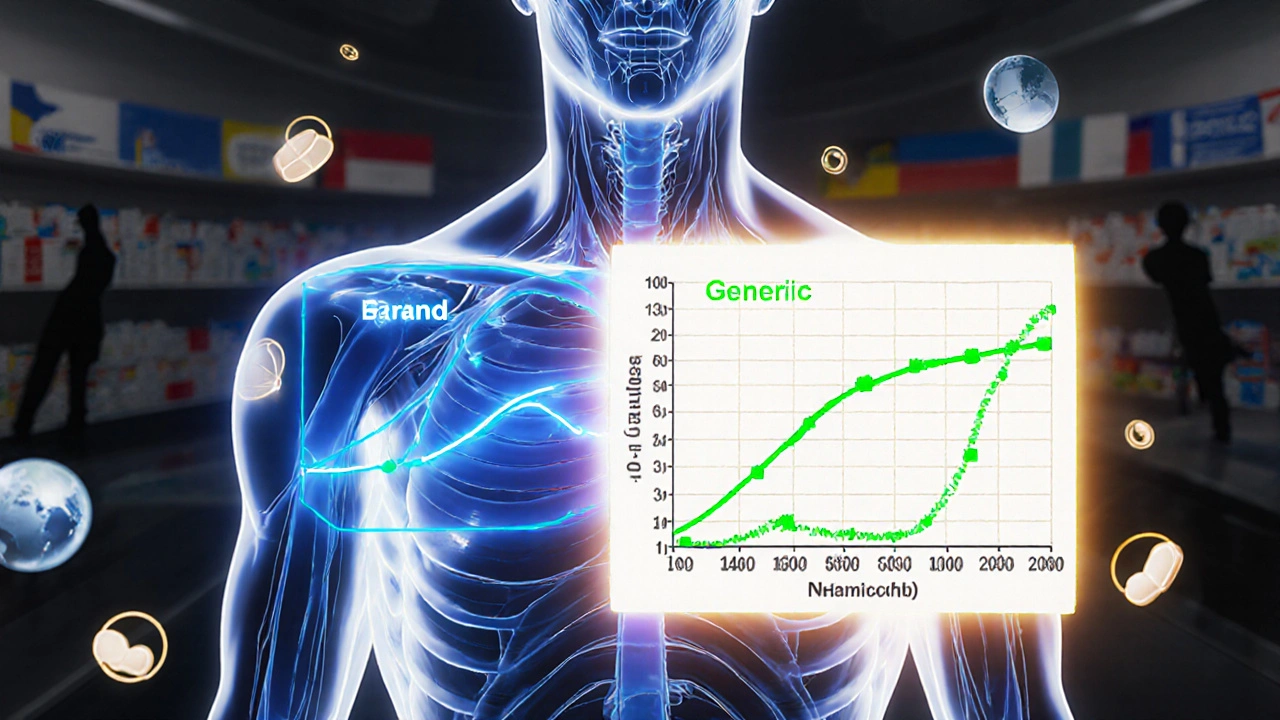
Many people think the 80-125% rule means generic drugs can contain anywhere from 80% to 125% of the active ingredient compared to the brand name. That’s wrong. A generic tablet doesn’t have 80% of the drug-it has nearly the same amount, usually 95-105%. The rule doesn’t apply to the amount of drug in the tablet. It applies to how your body absorbs it.
The 80-125% rule is a statistical gatekeeper. For two drugs to be considered bioequivalent, the 90% confidence interval of the ratio of their geometric means for two key measurements-AUC and Cmax-must fall entirely within that range. AUC (Area Under the Curve) tells you how much of the drug your body is exposed to over time. Cmax (maximum concentration) tells you how fast it gets absorbed. Both matter.
Why 90% confidence interval? Because regulators accept a 10% total risk of error-5% on each end. If the entire interval sits between 80% and 125%, it means there’s a 90% chance the difference in absorption between the generic and brand drug is small enough to be clinically meaningless. This isn’t a guess. It’s based on decades of post-marketing data showing that generics approved under this rule perform just as well in patients.
Why Logarithms and Geometric Means?
Drug concentrations in the blood don’t follow a normal bell curve. They follow a log-normal distribution. That means the data is skewed-most values cluster near the lower end, with a long tail toward higher concentrations. If you used regular averages (arithmetic means), you’d get misleading results.
So scientists take the natural logarithm of AUC and Cmax values. On this transformed scale, a 20% difference becomes symmetric: 80% equals -0.2231, and 125% equals +0.2231. This makes the math work properly. After analysis, they convert the results back to percentages. That’s why the rule looks like a simple range, but behind it is complex, well-tested statistics.
How Bioequivalence Studies Work
Before a generic drug is approved, it goes through a clinical study with 24 to 36 healthy volunteers. These aren’t patients-they’re people with no major health issues. Each volunteer takes both the brand-name drug and the generic version, in random order, with a washout period in between. Blood samples are taken over 24-72 hours to measure drug levels.
The study design is called a crossover trial. It reduces variability because each person acts as their own control. After collecting all the data, statisticians calculate the geometric mean ratio of AUC and Cmax between the two drugs. Then they build the 90% confidence interval. Both parameters must pass the 80-125% test. If one fails, the generic is rejected.
For drugs with high variability-like warfarin or certain epilepsy meds-the standard range might not be strict enough. In those cases, regulators use scaled average bioequivalence (SABE). This lets the acceptance range widen based on how much the brand drug’s absorption varies from person to person. For example, if the within-subject variability is over 30%, the range can stretch to 69.84-143.19% for Cmax. This isn’t a loophole-it’s a smarter, science-based adjustment.

When the Rule Isn’t Enough
Not all drugs play nice with the 80-125% rule. Narrow therapeutic index (NTI) drugs are the biggest concern. These are medicines where even a small change in blood level can cause harm or treatment failure. Think levothyroxine for thyroid disease, warfarin for blood thinning, or phenytoin for seizures.
In 2022, the FDA proposed tighter limits of 90-111% for NTI drugs. Some countries already use this. The European Medicines Agency has also tightened standards for certain drugs. Why? Because a 20% difference in exposure might be fine for an antibiotic, but dangerous for a drug that controls seizures or prevents strokes.
Even then, the rule doesn’t catch everything. Two drugs can be bioequivalent but still cause different side effects due to inactive ingredients-fillers, dyes, or coatings. That’s why some patients report issues with generics. But studies show these problems are rare and usually tied to formulation differences, not bioequivalence failure. A 2020 FDA analysis of over 2,000 generic approvals found only 0.34% needed label changes after market release due to safety concerns.
Why This Rule Exists
Before the 80-125% rule, generic drug approval was messy. Some countries required clinical trials proving a generic worked as well as the brand-expensive, slow, and unethical when the active ingredient was identical. The Hatch-Waxman Act of 1984 in the U.S. changed that. It allowed manufacturers to prove bioequivalence instead of redoing full clinical trials.
The 80-125% rule became the global gold standard because it’s practical. The FDA, EMA, WHO, Health Canada, and China’s NMPA all use it. That’s why a generic made in India can be sold in Australia, the U.S., or Germany without retesting. It’s one of the few areas where global health policy is truly aligned.
It’s also why generics now make up 90% of prescriptions in the U.S. but only 23% of drug spending. The rule saved billions. It made life-saving drugs affordable. And it didn’t sacrifice safety. Over 14,000 generic drugs have been approved under this rule in the U.S. alone. Post-marketing surveillance shows no spike in adverse events.

Common Misconceptions
Despite decades of use, confusion still runs deep. A 2022 survey found 63% of community pharmacists thought the 80-125% rule meant generics had less active ingredient. That’s not true. The active ingredient is tightly controlled-usually within 95-105% of the label claim, same as brand drugs.
Patients worry too. Online forums are full of stories: “My generic seizure meds don’t work.” “I had side effects after switching.” But when experts dig into those cases, fewer than 1 in 6 are actually linked to bioequivalence. More often, it’s a change in filler, a different release profile, or even psychological factors.
And yes, the rule was born from expert opinion, not clinical trials. In 1986, regulators asked: “What difference in absorption would actually hurt a patient?” Based on clinical experience, they settled on 20%. No randomized trial proved 80-125% was perfect. But 40 years of real-world use have proven it works.
What’s Next?
The system isn’t frozen. The FDA is investing $15 million over the next few years to explore model-informed bioequivalence-using computer simulations to predict how a drug behaves without testing in humans. For complex drugs like inhalers, topical creams, or extended-release pills, the 80-125% rule doesn’t always fit. That’s why the Complex Generics Initiative was launched in 2018.
Future standards may even account for genetics. Some people metabolize drugs faster due to their DNA. In the next decade, bioequivalence might need to consider pharmacogenomics. But for now, the 80-125% rule remains the backbone of generic drug approval worldwide.
It’s not perfect. But it’s the best tool we have. And it’s worked-quietly, reliably-for nearly half a century.
Is the 80-125% rule about how much active drug is in a generic pill?
No. The 80-125% rule does not refer to the amount of active ingredient in the tablet. Generic pills must contain the same amount of active drug as the brand name-typically within 95-105% of the label claim. The rule applies to pharmacokinetic measurements (AUC and Cmax) from clinical studies, which track how your body absorbs and processes the drug after ingestion.
Why is a 90% confidence interval used instead of a 95% one?
The 90% confidence interval is used because it allows for a 5% error margin on each side of the range (80% and 125%), totaling 10% overall risk. This is different from traditional hypothesis testing, which uses 95% CI to detect differences. Here, the goal is to prove similarity, not difference. The 90% CI strikes a balance between statistical rigor and practicality for regulatory decision-making.
Do all generic drugs need to pass the 80-125% rule?
Almost all immediate-release oral generics do. But exceptions exist. Highly variable drugs (like warfarin) may use scaled bioequivalence with wider limits. Narrow therapeutic index drugs (like levothyroxine) may require tighter limits of 90-111%. Complex formulations-such as inhalers, topical creams, or extended-release tablets-may need alternative testing methods approved by regulators.
Can a generic drug be approved even if it doesn’t meet the 80-125% rule?
Only under specific exceptions. For drugs with high variability, regulators allow scaled average bioequivalence (SABE), which expands the range based on the reference product’s variability. For some complex products, in vitro tests or pharmacokinetic modeling may replace in vivo studies. But if a drug fails the standard 80-125% rule and doesn’t qualify for an exception, it cannot be approved as a generic.
Why do some patients say their generic meds don’t work as well?
Most reports aren’t due to bioequivalence failure. Differences in inactive ingredients-fillers, dyes, coatings-can affect how quickly a pill dissolves or how it’s absorbed. Some patients are sensitive to these changes. Psychological factors also play a role: if someone believes generics are inferior, they may perceive reduced effectiveness. Post-marketing data shows that fewer than 20% of reported issues are linked to bioequivalence, and most are resolved without switching back.



Gabe Solack
November 17, 2025 AT 21:35Man, I used to think generics were just cheap knockoffs until I read this. The 80-125% rule is wild when you think about it - it’s not about how much drug is in the pill, it’s about how your body handles it. 🤯 I’ve been on generic levothyroxine for years and never had an issue. Science is cool like that.
Shilpi Tiwari
November 18, 2025 AT 20:24Log-normal distribution of pharmacokinetic parameters necessitates geometric mean ratios for accurate estimation of bioequivalence, as arithmetic means are inherently biased under right-skewed data. The 90% CI constraint is statistically optimized for regulatory decision-making under the assumption of multiplicative variability - a paradigm established by the FDA’s 1992 guidance and globally harmonized via ICH guidelines. Fascinating how empirical pragmatism trumped theoretical purity here.
Denny Sucipto
November 19, 2025 AT 09:52I used to freak out switching to generics, honestly. My grandma would always say, 'That ain't the real stuff.' But after reading this? I get it now. It's not about the pill - it's about your body doing the same thing with it. No magic, just math. And yeah, sometimes the filler makes your stomach act up, but that’s not the drug’s fault. You good, man. You good.
Holly Powell
November 19, 2025 AT 23:43Let’s be real - the 80-125% rule is a regulatory compromise born of industry lobbying, not clinical necessity. The FDA’s 0.34% post-market failure rate is meaningless when you consider that adverse events are underreported by 90%. And don’t even get me started on how they ignore inter-individual pharmacogenomic variability. This isn’t science - it’s a cost-cutting illusion dressed in statistical jargon.
Emanuel Jalba
November 21, 2025 AT 13:00THEY’RE LYING TO YOU. 🚨 I switched to generic seizure meds and my seizures got worse. The FDA knows this. They’ve been covering it up for decades. Why do you think they changed the label to say 'may differ in inactive ingredients'? BECAUSE THEY’RE SCARED. This isn’t science - it’s corporate control. Wake up. 🌪️💊
Heidi R
November 23, 2025 AT 08:21That 90-111% for NTI drugs? Long overdue. Why are we still allowing 20% swings for anything that can kill you? This rule is a relic. Time to update it. Full stop.
Brenda Kuter
November 24, 2025 AT 07:39They’re testing this on healthy volunteers? But what about people with liver disease? Or diabetes? Or anxiety? They don’t even test on real patients. How is that ethical? This is just a lab experiment pretending to be medicine. I’m not taking anything made under these conditions. Ever.
Sarah Frey
November 25, 2025 AT 00:47Thank you for this clear, well-researched breakdown. It’s refreshing to see such a nuanced topic explained without sensationalism. The distinction between active ingredient concentration and pharmacokinetic absorption is critical, and too often lost in public discourse. The 80-125% rule, while imperfect, remains a robust and globally validated framework. The real challenge lies in patient education - not regulation.
Katelyn Sykes
November 26, 2025 AT 18:02So the rule isn't about how much drug is in the pill but how your body uses it? That's wild. I always thought generics were just weaker. Turns out I was wrong. And the log-normal thing? Mind blown. We need more posts like this. Seriously. The world needs to know this stuff. 💪
Yash Nair
November 27, 2025 AT 15:55USA and EU get to decide what works for everyone? What about India? We make 40% of the world’s generics. Your rules are fine but dont ignore us. We have better QC than you think. And dont call it 'global standard' like its your invention. We helped build it too.
Bailey Sheppard
November 28, 2025 AT 09:02Really appreciate how you broke this down. I used to be skeptical about generics too - worried they wouldn’t work. But now I see it’s not about brand names or price tags, it’s about how the body processes the drug. And honestly? That’s all that matters. If it gets in your system the same way, you’re good. Thanks for the clarity.
Girish Pai
November 29, 2025 AT 13:29India exports 1000+ generic drugs to USA every year. Our labs are ISO certified. Your regulators trust us because we deliver. You think 80-125% is loose? Try manufacturing 500M tablets with 99.9% consistency. That’s real science. Not your lab experiments.
Kristi Joy
November 29, 2025 AT 21:57If you’ve ever been told your generic didn’t work, I get it. It’s scary to switch. But most of the time, it’s not the drug - it’s the filler, or your brain expecting it to be different. You’re not alone. Talk to your pharmacist. Ask about the formulation. Sometimes a small change makes all the difference. You’re not crazy. You’re just paying attention.
Hal Nicholas
November 30, 2025 AT 16:04Oh wow. Another one of these 'trust the system' rants. You really believe this 80-125% thing is foolproof? I’ve seen people die because their generic blood thinner didn’t work. You think the FDA cares? They get paid to approve, not to protect. Wake up. This isn’t medicine - it’s a numbers game.
Louie Amour
December 2, 2025 AT 08:41Let me guess - you’re one of those people who thinks 'bioequivalence' means 'same effect'. No. It means 'statistically similar within a 20% margin of error'. That’s not the same. That’s a gamble. And you’re the one who’ll take the pill. Good luck with that.